Methylmalonic aciduria
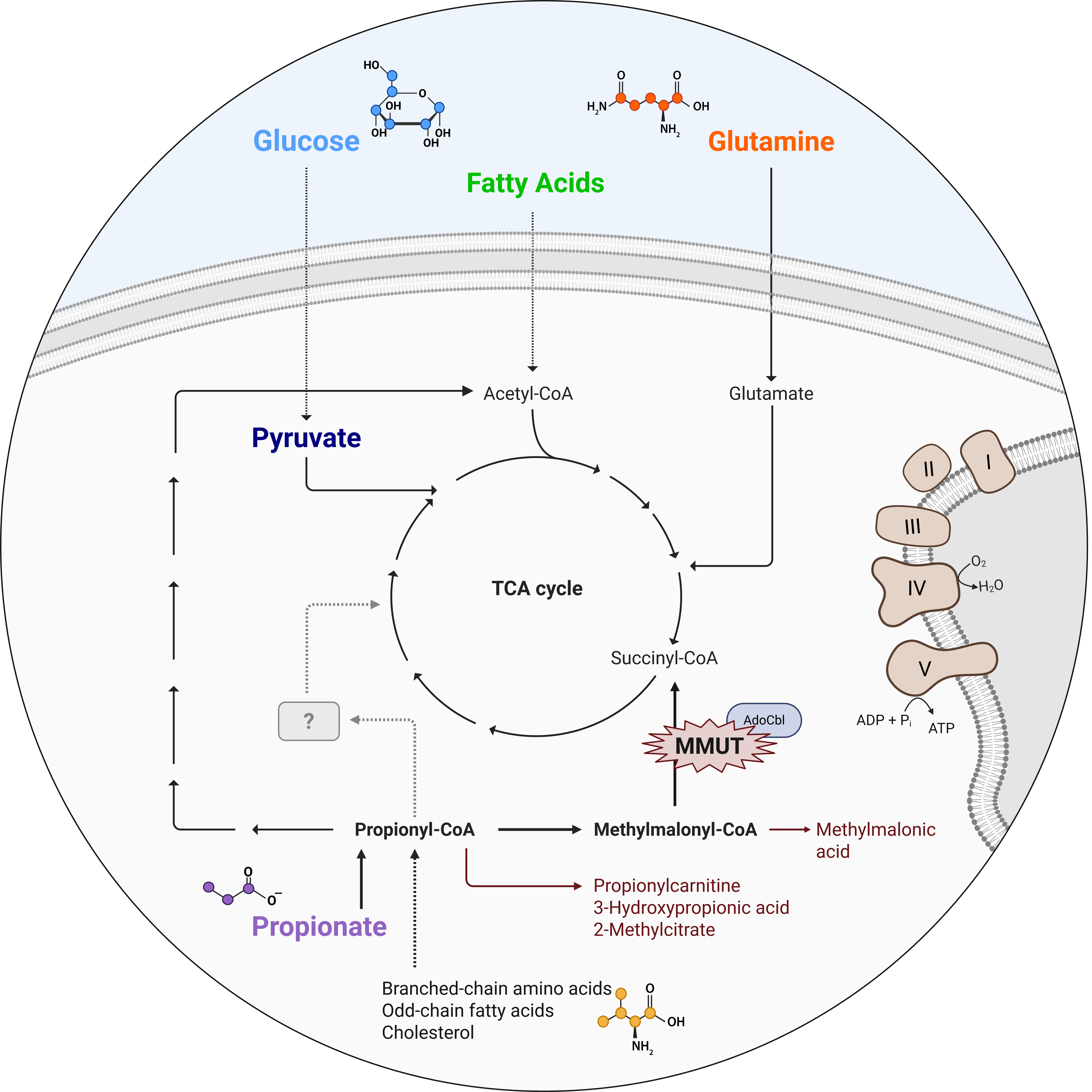
Methylmalonic aciduria (MMA) is a rare inherited metabolic disorder caused by deficiency of the mitochondrial enzyme methylmalonyl-CoA mutase (MMUT; MIM #251000) or the synthesis of its cofactor adenosylcobalamin. MMA is frequently life-threatening in the newborn or early childhood period and has devastating long-term consequences. Current therapeutic approaches are insufficient. Patients still experience metabolic crises, neurologic symptoms, progressive kidney failure and blindness. Normally, MMUT converts branched-chain amino acids, odd-chain fatty acids and the cholesterol side-chain into succinyl-CoA, an essential metabolite of the energy generating tricarboxylic acid (TCA) cycle. In MMA, deficiency of MMUT leads to decreased energy generation via the TCA cycle and accumulation of toxic metabolites. A promising treatment strategy may involve restoring mitochondrial energy generation. However, the most effective targets for this approach are unknown, and it remains uncertain whether they can be applied to the whole body. We are addressing these issues by exploring metabolic adaptations in MMA, including altered energy sources, pathway rerouting and impact on mitochondrial function.