LoF mutations - Methylmalonic aciduria
Autosomal recessive methylmalonic aciduria (MMA) is caused by loss of function of the mitochondrial enzyme methylmalonyl-CoA mutase (MMUT). The ensuing accumulation of toxic metabolites and reactive oxygen species contribute to mitochondrial dysfunction. Symptoms include acute metabolic decompensation leading to early death, chronic kidney disease with renal failure, and neurological sequelae. Although 70% of the MMA patients harbor at least one missense allele, most mutant proteins retain low residual activity. A small increase in activity may thus alleviate the disease. Through a high content small molecules in vitro screen (using repurposing libraries, natural compounds, and DNA-encoded libraries in MMUT-reporter HEK cells), we aim to identify compounds that increase protein stability and retention either directly or by inhibiting MUT ubiquitylation. Moreover, we will test the effects of bortezomib (inhibitor of 26S proteasome) on mutant MMUT degradation. Top candidates will be validated by testing mitochondrial dysfunction in invitro (MMutflox mouse kidney cells, patient derived iPS cells), and in vivo in zebrafish (MMUT-deficiency induced with CRISPR-Cas9) and mouse models (MMutKI/KO model for MMA). We will evaluate MMUT protein stability, mitochondrial function, accumulation of toxic metabolites, cell survival, and in vivo functional rescue (zebrafish and mouse).
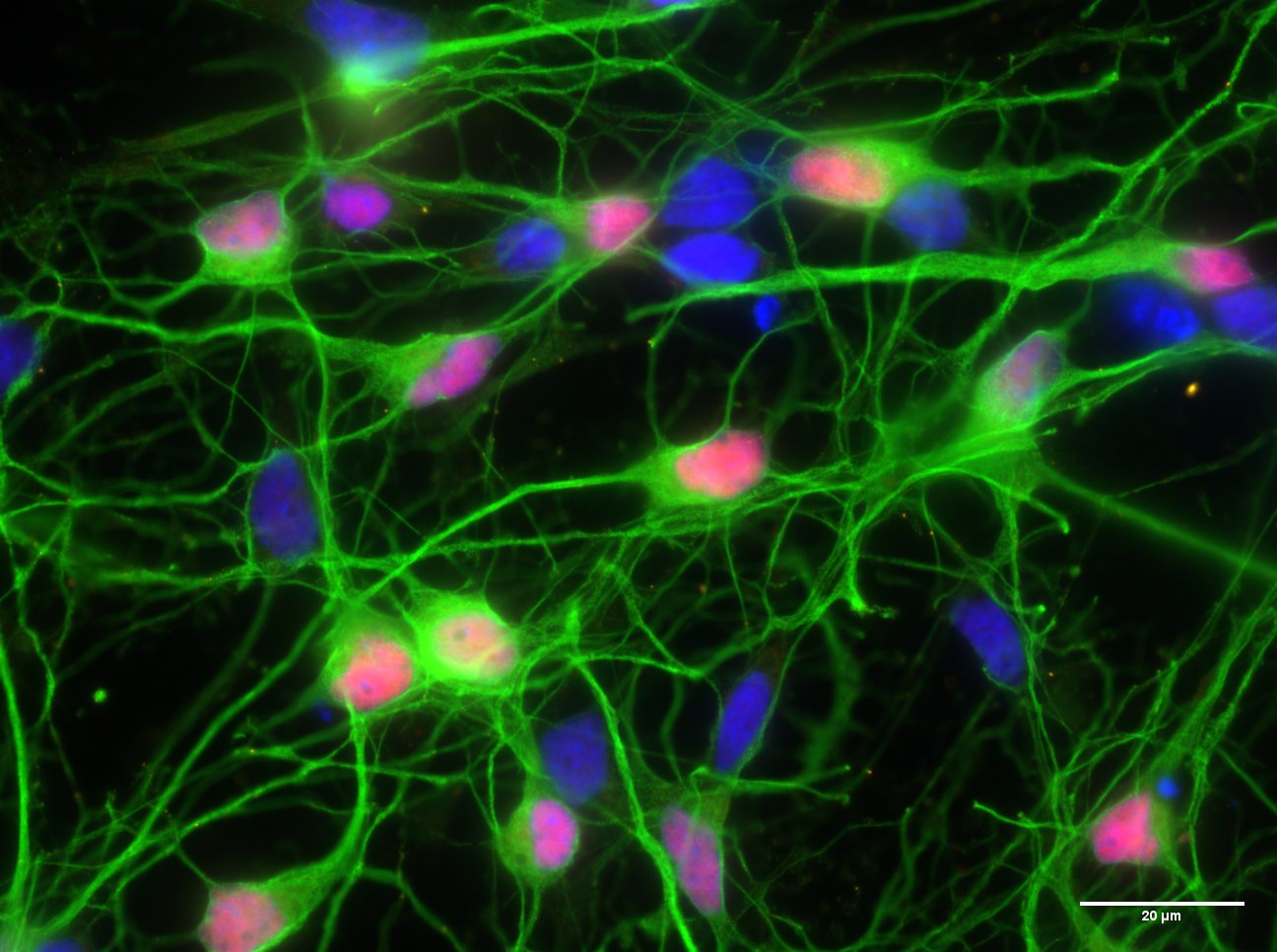
Figure 1. Neurons grown in the lab and stained for DNA (DAPI, blue), a transcription factor (TBR1, red) and the cytoskeleton (TUJ1, green).